Deep learning for transcription factors binding sites predictions
Publication (in preparation): maxATAC-v2 infers nucleosome positions from single-cell ATAC-seq training data for improved genome-wide transcription factor binding site prediction
Poster presentations:
- Senior Capstone 2025: Nucleosome positions improve cell-type specific transcription factor binding site predictions using deep-learning based maxATAC-v2
- CCHMC Immunology Retreat 2023: Improved transcription factor binding site predictions and benchmarking using deep-learning based maxATAC-v2 models
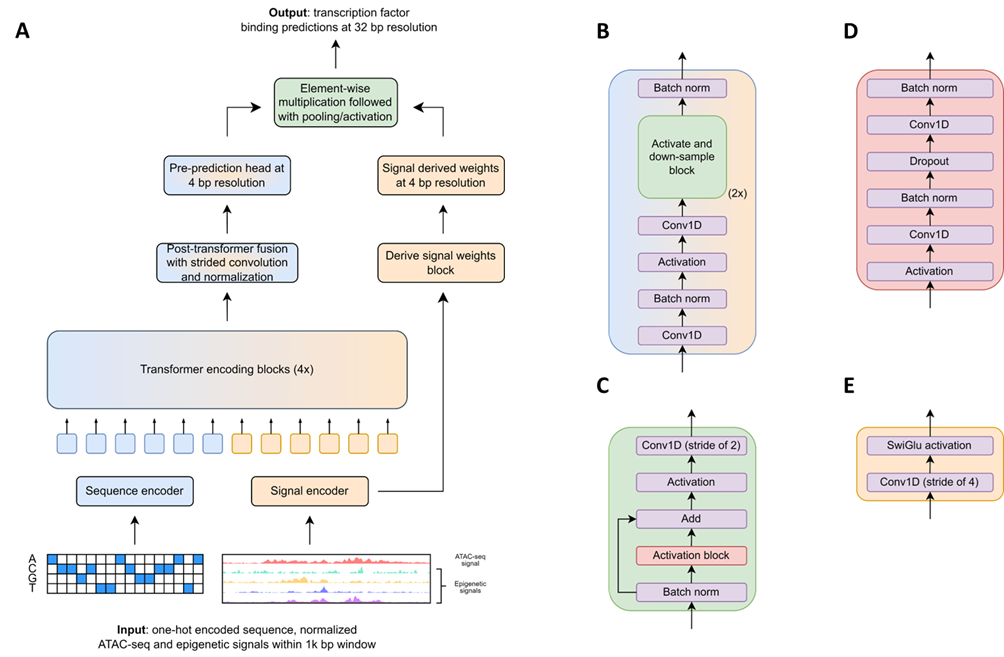
Transcription factors (TF) regulate gene expression by binding to specific DNA sequences called motifs. However, the presence of motifs does not guarantee TF binding in vivo, due to other epigenetics factors such as chromatin accessibility, DNA methylation marks, … In this project, we investigate the use of deep learning to predict cell-specific TF binding across 127 human TFs, utilizing inputs from the reference DNA sequence and ATAC-seq signal. The work utilizes the transformer architecture to integrate multimodal epigenetic inputs and predict binding at 32-bp resolution.
My contributions include building the prototype transformer, benchmarking its performance on held-out hematopoietic stem cells, curating and cross-correlating bulk and single-cell ATAC-seq data, and leading analyses for attention matrix visualizations for interpretability.
My utmost gratitude to Dr. Matthew Weirauch and Dr. Emily Miraldi for allowing me to work on the project!